

Introduction
On August 25th, the Office of Pharmaceutical Quality (OPQ), part of the FDA’s Center for Drug Evaluation and Research (CDER) released its fourth annual Report on the State of Pharmaceutical Quality, covering fiscal year 2021. Generally speaking, the OPQ is tasked with ensuring that legally marketed drugs in the US are safe, effective, and meet quality standards, and the annual report characterizes the current state of drug and site quality to inform stakeholders, such as consumers and patients, about the current quality of the US drug supply. The report does conclude that the current state of quality in the pharmaceutical industry in the US is high, but points to the need for and expectation of continual improvement.
Aside from presenting key data derived from extensive OPQ research to communicate the state of pharmaceutical product and manufacturing site quality, the report also highlights two initiatives aimed at advancing pharmaceutical quality: the New Inspection Protocol Project (NIPP) and Quality Management Maturity (QMM). These initiatives look to advance pharmaceutical quality and quality system maturity, which industry experts have long criticized as lagging behind other regulated industries such as medical devices, by improving the data produced by inspections and assessments to generate a more robust understanding of site quality, which will aid the FDA in making better, more informed, and timelier decisions as well as driving pharmaceutical manufacturers to continually improve.
Addressing the Challenges Brought on by the COVID-19 Pandemic
The 2021 fiscal year was particularly interesting and challenging for the Agency as it was still rocked by the limitations in site access and travel restrictions that were brought on by the COVID-19 pandemic. This, of course, pushed the Agency into the proliferation and advancement of otherwise unconventional methods of inspections, with many of the methods developed now being considered as a mainstay for future inspections. Alternate tools, the report explains, enabled the FDA to assess sites that would otherwise have been out of reach, such as the MRA, or Mutual Recognition Agreement, which is a program that enables the FDA to receive and rely upon inspection reports from partner agencies. The report states that by using inspection reports from MRA partners, the Agency was able to review and classify 139 site inspections in 18 MRA partner countries and six other countries.
The Agency also used its authority granted by the DF&C Act to request records and information in lieu of or in advance of inspections to assess cGMP compliance in areas where the Agency would otherwise not be able to assess. During FY21, 288 surveillance systems-based assessments were conducted by the FDA using such methods and assessing records that were supplied by sites in which an onsite inspection could not be conducted. As a testament to efficacy, 21 Import Alerts were generated as a result of these assessments. These assessments, since dubbed Remote Regulatory Assessments, or RRAs, have since been codified as of this July in draft guidance to outline their future use.
The 17-page draft guidance is in question-and-answer format and covers important topics regarding RRAs such as who would be subject to an RRA, whether RRAs are replacing other established means of obtaining information outside of inspections, whether the RRA counts as an inspection, and expectations regarding RRAs such as how the FDA requests them and what consequences manufacturers face if they decline participation.
The new proposed guidance “lays out how we intend to leverage this tool [RRAs] to advance our mission as we further incorporate modernized approaches to protecting public health” said FDA Commissioner Robert Califf and Judith McMeekin, associate commissioner for regulatory affairs, explaining how the remote regulatory assessment tools were not intended to replace onsite inspections, but rather to work in conjunction with them.
Further advancement is pharmaceutical product and site quality during FY21 is evidenced in the report’s section covering the FDA’s list of Essential Medicines, Medical Countermeasures, and Critical Inputs, otherwise known as EM, as it will be referred to throughout the remainder of this article. The EM list was created in response to Executive Order 13944 in October of 2020 in an effort to further protect Americans from outbreaks of infectious diseases, such as COVID-19, and chemical, biological, radiological, or nuclear threats. The Executive Order aimed at reducing shortages in the supply chain of critical products to combat these threats.
The EM answers this need by listing approximately 1,100 sites that manufacture at least one of the listed 227 drug and biological essential medicines and countermeasures, including analgesics, antivirals, anticoagulants, antihypertensives, and antimicrobials. Of note, the report also concluded that the median Site Inspection Score (SIS) for manufacturers who are listed in the EM is significantly higher than the SIS for non-EM manufacturers, suggesting that EM products also have a higher degree of compliance to cGMP than products manufactured by firms that are not listed. A troubling fact when the implications are assessed along with data presented later in the report regarding the relationship between recalls and inspections all ultimately bring to light the notion that firms, when not under the watchful eye of the Agency, also tend to have lower compliance to regulatory quality standards.
By the Numbers
The FDA evaluates mandatory and voluntary post-market quality reports. Below is a chart highlighting the reports that were received and evaluated by the Agency during FY21, with the numbers being very similar to those of FY20. The CARES Act, signed into law in March of 2020 to address supply chain security and shortages of critical drugs and promote innovation by fostering collaboration between government and the private sector, requires increased reporting, which will enable the Agency to normalize these reports by the amount of each product manufactured for commercial distribution, which should also improve future evaluation to better understand the impact and magnitude of the PDQs.

Further, import alerts were placed on 49 sites for refusing inspections, refusing record requests, non-complaint laboratory testing, and non-compliant findings from inspections and record requests during FY21, with the largest number of import alerts being issued to firms in China and Latin America and with all non-compliant laboratory testing alerts being issued to manufacturers of hand sanitizers, illustrating the proliferation of these types of products during the pandemic and the action by the Agency to protect American consumers.
The number of total recalls for Class I devices increased for the second year in a row, also due largely to hand sanitizer products removed from the market that contained methanol. Class II recalls continue to not show trends, being primarily event-driven, and Class III recalls, likewise, continue to be steady.
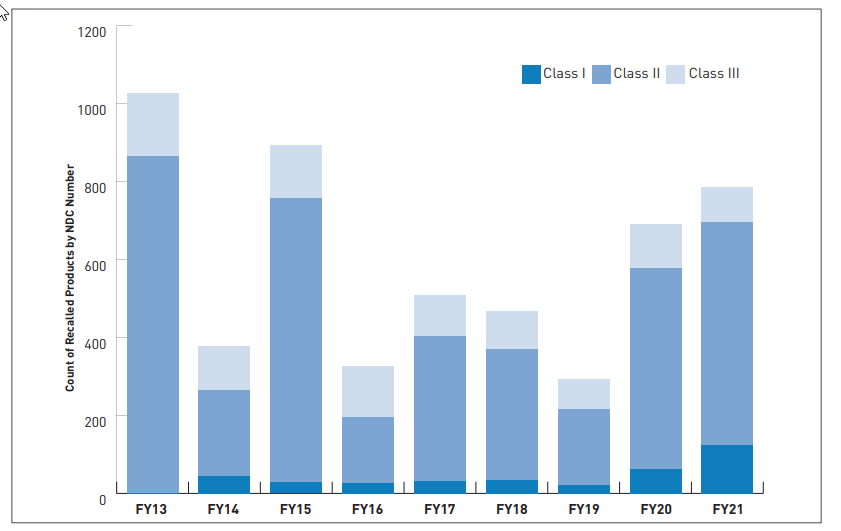
To understand the relationship between inspections and recalls, which in turn is needed to understand the efficacy of inspections in protecting American consumers as well as manufacturers’ ability to comply with cGMP and maintain the production of high-quality and safe products, the report publishes conclusions from FY17-21 which highlighted the following:
- There are more Class II recalls in the 12 months following surveillance inspections than what occurs outside of the 12-month window
- There are more Class III recalls in the 6 months following surveillance inspections than what occurs outside of the 6-month window
- Class I recalls are generally associated with final Official Action Indicated (OAI) classification and an initial OAI classification that was reclassified to VAI (Voluntary Action Indicated) after the resolution of violations.
While it’s important to note that the statistics do illustrate the efficacy of inspections in removing potentially dangerous pharmaceutical products from the market and protecting American consumers, it also causes a pause to highlight the importance of manufacturers’ understanding their obligations to ensure that products adhere to specifications and cGMP. Without these inspections, the data would suggest, these non-compliant products would still be on the market and bad actors would go unrecognized.
New Initiatives to Advance Pharmaceutical Quality
The New Inspection Protocol Project (NIPP) seeks to modernize the FDA’s inspection program by improving how data from surveillance and pre-approval inspections are recorded, assessed, and reported, the report states, noting that the methods have been used in sterile applications since 2018 to improve consistency of inspection and to better catch issues that could lead to drug shortages, and that the Agency is currently developing and deploying protocols in non-sterile applications as well. The collection of structured data through questions and responses on inspections is said to enable more efficient and robust analytics and drive data-driven decisions, facilitating data mining processes to find anomalies, patterns, and correlations between data collected from inspection sites. Adopting such Industry 4.0 practices will aid the Agency in gaining insights to understand when pre-approval inspections are warranted as well as gathering data to help understand when proactive measures may be needed to mitigate systemic issues.
To better understand how the NIPP is helping the Agency to promote better quality in the industry, it was originally stated that the goal was to expand an investigator’s focus beyond identifying cGMP violations and instead to enable the investigator to be able to evaluate the overall state of a facility’s quality systems and operations – a move that highlights the needs of manufacturers in the regulated space to think beyond merely meeting regulatory requirements and instead focusing on building mature quality systems to ensure the consistent delivery of high-quality products, removing barriers to supply-chain disruption. While the protocols are not publicly available, we know that the Agency uses several high-tech tools and methods to achieve its goals using the protocols such as tablets pre-loaded with protocols used for rating facilities during inspections and advanced algorithms to identify high-risk sites. Both manufacturers and inspectors will need to be ready to think beyond regulatory compliance and shift their focus toward a constant state of quality, which leads to the next initiative highlighted in the report.
In an effort to better understand Quality Management Maturity (QMM), two pilot initiatives were started by the FDA to objectively measure quality system maturity levels, with the pilot programs being prompted by the understanding of the importance of a robust, mature quality system in the industry to protect consumers and meet market needs, noting that 62% of drugs that went into shortage between 2013 and 2017 did so as a result of quality-related problems such as sub-standard manufacturing sites and processes or defects in the final products. The time and costs associated with remediation of such quality problems inevitably lead to shortage, and, conversely, a mature quality system increases efficiency and reduces risk of shortage in the supply chain. QMM is described as a state attained by having consistent, reliable, and robust business processes to achieve quality objectives and promote continual improvement, and, aside from being able to consistently and objectively measure quality management maturity, the Agency is seeking methods of rewarding those firms that have adopted mature quality management practices.
The pilot programs utilize unique methods of collecting information such as self-surveys, interviews based on an assessment rubric, and focused presentations delivered by subject sites to try to balance the burden of collecting information while also achieving accurate QMM scores. The assessments focused on proactive management of product availability risks, effective application of quality risk management throughout the business, investments into digitization, and the use of advanced analytic tools. The FDA will continue to mature the assessment methods from the pilot programs in collaboration with stakeholders to ensure that the program will meet objectives in being able to uniformly and objectively assess the maturity of firms’ quality systems without placing an undue burden on anyone as information is collected and assessed.
Conclusions
While recall and PQD data illustrate that the state of quality is fairly strong in the pharmaceutical industry, it is also evident that compliance to cGMP is far from uniform and that inspections continue to be a necessary control to limit poor quality products from entering the US market. Further, poor quality and non-compliance with regulations have continued to cause shortages in already constrained supply chains, putting Americans at risk both of poor quality and short supply of life-saving products.
While methods of conducting inspections remotely and collecting data through requests for records are improving and helping to keep non-compliant firms in check, the ability to process and analyze this data is lagging behind other industries or lofty Industry 4.0 ideals, but new initiatives are helping to make better use of data to predict when pre-approval inspections are needed and to take mitigating actions to address systemic issues.
Firms who are manufacturing or distributing products in the industry have an obligation to follow regulatory requirements and implement cGMP to ensure the safety of end users and maintain a consistent supply of their products to the market and having a robust quality system is certainly a key factor in ensuring that those obligations are met. The FDA, in its commitment to the continual advancement of quality in the industry, is seeking to improve methods of objectively assessing quality management maturity through QMM assessment pilot programs, which will surely lead to more consistent compliance in the future, as well as the ability to better recognize and reward firms who build mature quality systems to ensure the safety and consistent supply of their products.